First. Samuli Ollila wrote a preliminary manuscript demonstrating that the widely used Berger force field appeared to have some trouble correctly reproducing the lipid head group conformations measured in NMR experiments. Also responses to changes in hydration levels, ion concentrations and cholesterol content differed between experiments and simulations with the Berger model. Other force fields were not tested.
Then. During the last two weeks data for Stockholm-, Kukol-, OPLS-AA-, and the unpublished Maciejewski–Rog force fields have been made available by commenters (thanks guys!) of this blog.
{kind=link}
{kind=link}
{kind=link}
Now. The plots below summarize the current status of our knowledge:
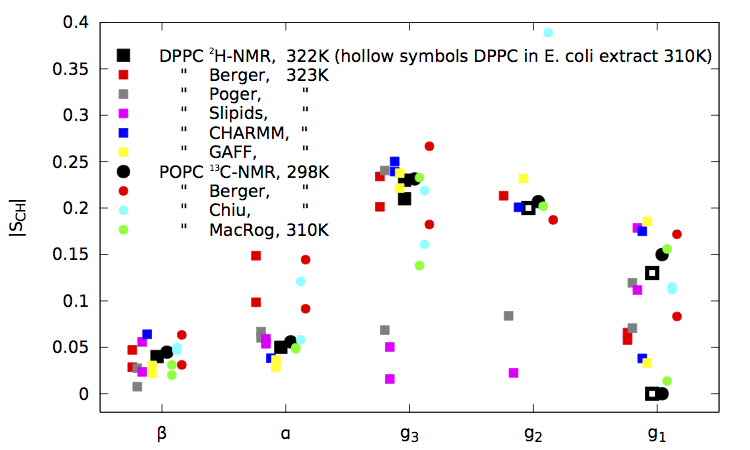 |
| Figure 1. NMR results, original Berger data, contributions obtained through the blog so far, and the GAFF and CHARMM results taken from the publications for pure fully hydrated bilayers. (Figure last updated on October 26th 2013.) |
 |
| Figure 2. Data of systems containing NaCl for the Kukol and OPLS-AA force fields. (For these we currently have no data of pure fully hydrated bilayers and thus decided not include them in Figure 1.) |
It seems that in addition to the GAFF and CHARMM, which were known to be good, also the Maciejewski–Rog force field gives significantly better order parameters than the Berger model.
Next. As always, all contributions are warmly welcome. However, here are maybe the most critical things we should obtain next:
- Response of the good all-atom force fields (GAFF, CHARMM, Maciejewski–Rog) to changing conditions: hydration levels, ion concentrations, cholesterol content. If you have done simulations with these force fields and would be willing to either (1) share the input files for Gromacs or (2) report the order parameters here, it would be a great benefit for the project. (All contributors will become authors of the final paper, see the post On credits.)
Order parameters from Chiu et. al. united-atom force field. They have put effort in parametrizing headgroup torsions, thus this could be the united atom force field that gives a significant improvement over Berger. Here the input files seem to be available, but if ready trajectories already exist, posting order parameter data would be very useful.Update on Oct 8th: This FF has now also been tested, see comment by Samuli below. Figure 1 has been updated accordingly.- Comments, questions, criticism. There are errors in all manuscripts, and we would be very grateful if you would report should you have found one—no matter how small or big. For example, based on the feedback we have got outside this blog, there are people who have known for a long time that the headgroup is not well described in the Berger model. We would be very grateful if you could share your insights and, e.g., point out missing relevant literature in the manuscript. Another cool example of a very relevant contribution could be if an impartial NMR expert could provide us with their insight and write a post on the reliability of the experimental data used for the comparisons. But these are just examples. Please let us know if there is anything that you think we should have taken into account.
markus & Samuli