As discussed in
the previous post, the NMRlipids IV project concerning PE, PG and PS lipids was divided into two parts. The first part about the results from PS lipids
is approaching to the submission, while the second part about PE and PG lipids is in a more preliminary stage.
I have now started to assemble the contributed data for PE and PG lipids into
a manuscript in
a new GitHub repository. From now on, I will call this part of the project as
NMRlipids IVb. Current status of the manuscript and most important things to todo are summarized in this post.
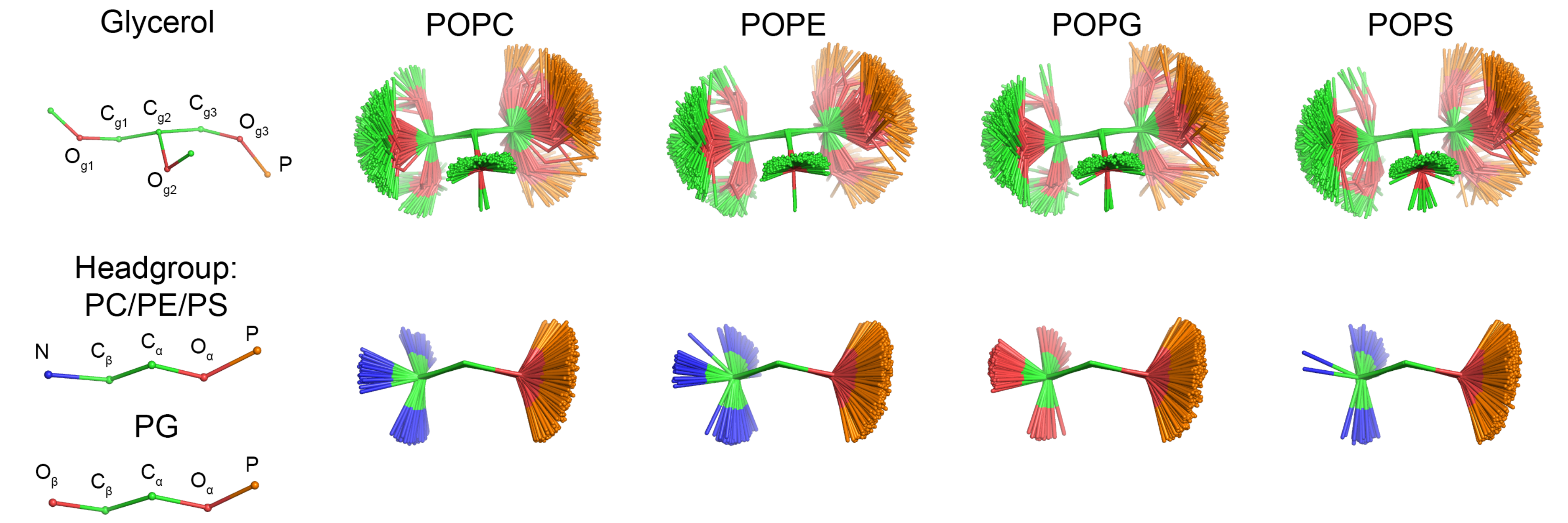
Headgroup and glycerol backbone order parameters from lipids with different headgroups in experiments
Experimental order parameters delivered by Tiago Ferreira with the information about the sign revealed that the 𝛽-carbon order parameter of PG lipids is positive, while other lipids have negative order parameter for this carbon (Fig. 1). Therefore, the structure of PG lipid seems to be distinct from PC and PE, in contrast to the conclusions from the order parameter data without sign information
in the previous post.
 |
| Figure 1: Experimental order parameters of the headgroup and glycerol backbone region C-H bonds from experiments. For literature references, see the manuscript draft. |
Headgroup and glycerol backbone order parameters of PE and PG lipids in simulations
The experimental information about order parameter signs enables a similar comparison between different simulation models and experiments for PE and PG lipids as done previously for PC and PS lipids in
NMRlipids I and
NMRlipids IV projects. This comparison (Fig. 2) and the results in
NMRlipids I and
NMRlipids IV projects suggest that the differences between PC, PE, PG, and PS headgroups are well captured in CHARMM36 simulations, although order parameters of all C-H bonds are not within the experimental error for any of the lipids. Therefore, the structural differences between lipid headgroups could be analyzed from the CHARMM36 simulations, perhaps in a similar way as done for the PS headgroup in the
NMRlipids IV project. The contributed data for PE and PG lipids from united atom simulations is not yet analyzed, partly because of
the lack of suitable programs for the automatic analysis of the data.
 |
| Figure 2: Headgroup and glycerol backbone order parameters from PE (left) and PG (right) lipid bilayers from experiments and different simulation models. For literature references, see the manuscript draft. |
Cation binding to lipid bilayers containing PE and PG lipids
Sodium binding to PE lipid bilayers
seems to be slightly weaker than in corresponding simulations for PC lipids, but experimental data is not available. For calcium binding to PE lipids, we do not have experimental nor simulation data yet.
As discussed in the
NMRlipids IV project, the counterion binding to negatively charged lipid bilayer is complicated to assess against experimental data. However,
the results from PC:PG mixtures from CHARMM36 and Slipids simulations indicate that sodium ions could be slightly overbinding to PG lipids, similarly to PS lipids in the
NMRlipids IV project. For calcium binding to PG containing lipid bilayers, we have data only
from CHARMM36 simulations and the conclusions are not yet fully clear.
ToDo:
- Analyze united atom simulations of PE lipids (GROMOS, CHARMM36ua, OPLS-UA, Berger, and GROMOS-CKP) listed in Table I in the manuscript with or without the new code for united atom data.
- Analyze structural differences between POPC, POPE, POPG and POPS headgroups from CHARMM36 simulations. The most reasonable first step would be probably to follow a similar analysis that was done for PS lipids in the NMRlipids IV project.
- Simulation data for PG lipids with different force fields would be useful. Currently, we have only CHARMM36 and Slipids.
- Simulation data from POPC:POPG (1:1) mixtures with sodium counterions and different CaCl2 concentrations (0-1M) from different force fields would be useful. Currently, we have results only from CHARMM36.





{kind=link}