First of all, as a response to the first ToDo issue in the previous post, two different codes to calculate the order parameters from united atom simulations have been contributed and also thoroughly discussed (see discussion in the previous post and buildH program). This enabled the analysis of previously contributed united atom simulations and the results are now added into the manuscript. Also, significant amount of new data has been contributed and we have now a good collection of force fields for both PE and PG lipids.
The data is still suggesting that the CHARMM36 simulations capture the essential differences between PC, PE, PS, and PG headgroups. A figure of structural ensembles of different headgroups from CHARMM36 simulations is now contributed.
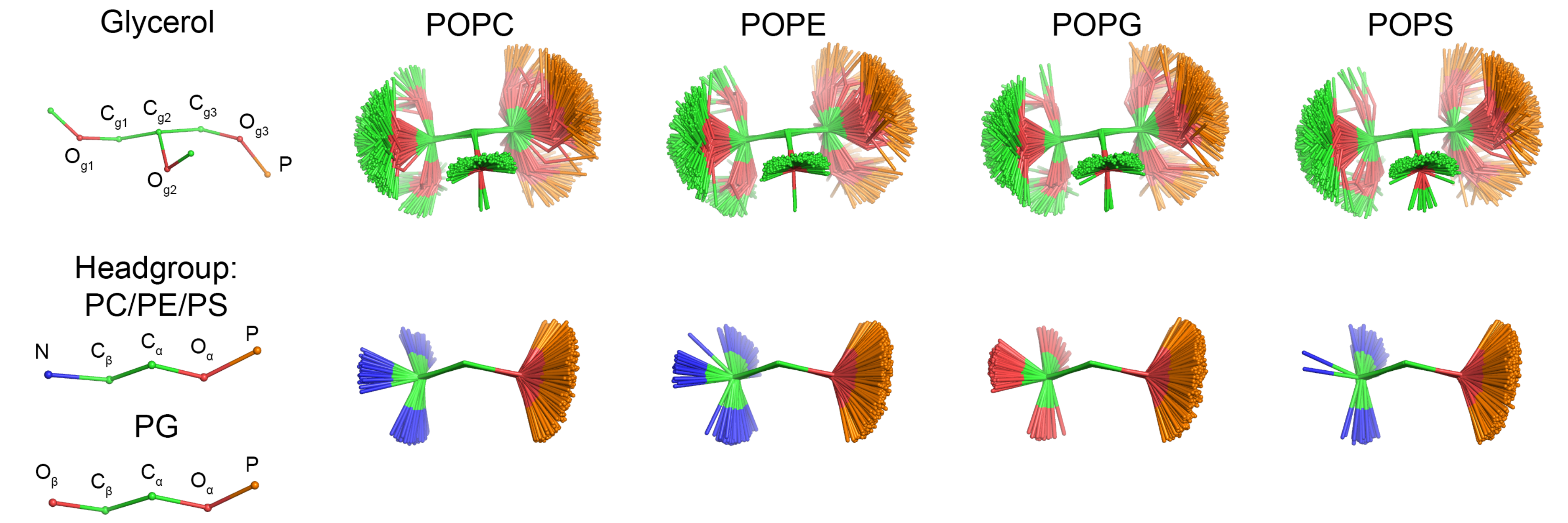 |
| Figure 1: Overlayed snapshots of a glycerol backbone and headgroup conformations of different headgroups from CHARMM36 simulations. Figure is made by Pavel Buslaev. |
For calcium-binding affinity to PG containing membranes we still need more data. The current data is not fully converged, and I think that we need longer CHARMM36 simulations with the recent NBfix correction for calcium and counterions. I have opened an issue in GitHub with more details for further discussion.
I have opened also other issues in GitHub. All the issues are important, but the most critical for the progress of the manuscript are the above mentioned analysis of structural differences between different lipid headgroups and additional data for PG-calcium interactions. In addition to the issues listed in GitHub, there are also Todo points in the manuscript (tex file) and SI (tex file). For example, we need citations for the force fields in tables I-III, and to finish the method sections for MD simulation and NMR experiments.
Dear all,
ReplyDeleteI have uploaded 8 independent runs of 500 ns DOPE in TIP3P, with velocities saved, onto zenodo:
https://doi.org/10.5281/zenodo.3557459
https://doi.org/10.5281/zenodo.3557451
https://doi.org/10.5281/zenodo.3557469
https://doi.org/10.5281/zenodo.3557473
https://doi.org/10.5281/zenodo.3557475
https://doi.org/10.5281/zenodo.3557478
https://doi.org/10.5281/zenodo.3557608
https://doi.org/10.5281/zenodo.3557669
These are probably not that necessary for the NMRlipidsIVb anymore, but I though it would not hurt to have them at least in the databank.
If there is anything missing, please send me an email.
Otto Schullian (MPIKG, PhD of M. Miettinen)
Thanks Otto. I have not yet added this into the manuscript because this data is indeed not necessary and I have a lot of things to do. However, it could be probably included if someone incorporates it into the databank and puts the results in figure 2 in the manuscript in the way that 8 independent simulations are presented in a reasonable way.
DeleteDear all,
ReplyDeletewith Paula Milan Rodriguez (Ph.D student under my supervision), we have performed some simulations of POPC/POPE (1:1) using the MacRog FF. The results are summed up in this document: https://www.dsimb.inserm.fr/~fuchs/project_Samuli/POPC_POPE/POPC_POPE_report.pdf
The itp files for POPC and POPE were initially downloaded from the SI of a paper in Data in Brief (10.1016/j.dib.2016.03.067). As posted earlier on the blog for POPS, we found some errors in the itp file of POPE (sn-1 and sn-2 switched thus it was OPPE, some atoms not connected by a bond, differents atoms with the same name). But we also found some errors in the itp file of POPC (some missing impropers leading to non planar systems for double bonds). Paula made some thorough testing, corrections and analyses of these files by doing also some simulations of pure POPC and pure POPE. Notably, she checked that the fixed POPC itp file reproduces the results of NMRlipidsI. Everything is described in this document: https://www.dsimb.inserm.fr/~fuchs/project_Samuli/POPC_POPE/report_results_comparison.pdf
Paula will post soon the links to the different trajectories on Zenodo (which contain the fixed itp files as well).
Best,
Patrick and Paula
Dear all,
DeleteHere are the links to the 3 Zenodo publications we carried out Patrick and I:
MacRog pure POPC:
https://zenodo.org/record/3725666#.Xn4K6vEo85k
MacRog pure POPE:
https://zenodo.org/record/3725670#.Xn4K1PEo85k
MacRog POPC/POPE 1:1:
https://zenodo.org/record/3725637#.Xn4K_vEo85k
As Patrick said, you can find the results and detailed informations for the POPC:POPE 1:1 simulation in this document :
https://www.dsimb.inserm.fr/~fuchs/project_Samuli/POPC_POPE/POPC_POPE_report.pdf
And for the pure POPC and pure POPE simulations in this other one:
https://www.dsimb.inserm.fr/~fuchs/project_Samuli/POPC_POPE/report_results_comparison.pdf
Best regards,
Paula Milan Rodriguez.
Hi,
DeleteI checked this zenodo for pure POPC (https://zenodo.org/record/3725666#.Xn4K6vEo85k). The trajectory and .gro therein seem to be for the mixed system but the tpr contains only POPC.
Best,
Hanne
Thanks Patrick and Paula, this data is now added into the manuscript. The PC headgroup seems to be affected more by PE in the MacRog simulation than in other models (Fig. 5 in the manuscript).
DeleteHi Hanne,
ReplyDeleteYou were right, there was an error in the uploading of the pure POPC simulation files (specifically the .gro and .xtc). I have generated a new version of the zenodo publication, you can check it here:
https://zenodo.org/record/3741793
Best,
PaulaMR
Thanks!
ReplyDeleteThe naming convention seems to be different from the MacRog runs already under nmrlipids zenodo. Do you happen to have the mapping and .def files for these atom names?
Best,
Hanne
Hi Hanne,
ReplyDeleteyes it is likely that our POPC have different atom names because the source is different (we got the initial POPC.itp from a publication [see our comments above and the pdf reports], while for NMRlipidsI Matti got the itp directly from the authors prior to publication). Also there were two C27 atoms, so one of them was renamed C28 by Paula. I can generate the mapping and def files. Actually, I'm developing a script that does this automatically. This is a good occasion to improve it before sharing it with the NMRlipids community.
Do you also need the mapping and def files for our MacRog POPE simulations ?
Best,
Patrick
Hi,
ReplyDeleteI don't need PE-files just now, but in general it would be awesome to have a script that does these automatically :) Maybe we could then start adding these mapping and .def files to the zenodo entries as a default.
Thanks!
-Hanne
All right, I will send you the mapping and def files for POPC. But I have one question before, it's about the mapping format regarding hydrogens. For example in the file https://github.com/NMRLipids/MATCH/blob/master/MAPPING/mappingPOPCmacrog.txt, for the g1 carbon we have :
ReplyDeleteM_G1_M C32
M_G1H1_M H322
M_G1H2_M H321
By inspecting a structure M_G1H1_M is pro-R and M_G1H2_M is pro-S.
By looking at another example, g3 :
M_G3_M CF
M_G3H1_M H2f
M_G3H2_M H1F
M_G3H1_M is pro-R and M_G3H2_M is pro-S.
Is this always the case for all hydrogens M_*H1_M is pro-R and M_*H2_M is pro-S ? Maybe it's written somewhere, but I didn't find that info.
If yes, I have to take that into account in my automatic mapping script. So far I didn't control which H is pro-R or pro-S. But I guess it's doable.
Thanks,
Patrick
Currently R/S hydrogens are not specified in the mapping file, i.e., they are completely random. It is not written anywhere, sorry about that.
DeleteHowever, it would be great if they were well defined. This would be useful in some situations. Therefore, it would be great if it would be possible to include this into your code without too much work. Naturally, we can also continue without defining them for now.
Thanks for your answer Samuli. After some thoughts, it's likely doable. Thanks to the functions we wrote for reconstructing Hs in buildH, I think we can easily distinguish between pro-R and pro-S while reading a structure. Furthermore, I checked that we can do graph matching by imposing attributes to some nodes (e.g. we can define an attribute "type" which can take a value "pro-R" or "pro-S" for each H in a CH2 group).
ReplyDeleteOf course this implementation will take some more time to be included into the automatic topology writer. I propose first to send to Hanne the .def and mapping files without taking this into account.
On the other hand, I start working on the inclusion of this implementation in my code.
Working first without distinguishing R and S hydrogens and then including this later on sounds good to me.
DeleteDear all,
ReplyDeleteI finally released the automatic lipid mapping writer, for now it's called autoLipMap: https://github.com/patrickfuchs/autoLipMap
If you want to use it, please read carefully the section "Known Issues".
I used it to generate the .def and mapping files for our simulations with Paula using a fixed POPC MacRog FF. I put the two files on my site :
- https://www.dsimb.inserm.fr/~fuchs/project_Samuli/POPC_POPE/automatic_mappingPOPCmacrog_fixed.txt
- https://www.dsimb.inserm.fr/~fuchs/project_Samuli/POPC_POPE/automatic_POPCmacrog_fixed.def
As we discussed, for now, there is no distinction between pro-R and pro-S.
Don't hesitate if you have questions.
Best,
Patrick
Thanks again Patrick, this seems highly useful!
DeleteI was testing it also to this data https://doi.org/10.5281/zenodo.3474862 that I needed to analyze for NMRlipids IVb. However, the code seems to detect too many bonds in this case. I have written a more detailed description into the issue: https://github.com/patrickfuchs/autoLipMap/issues/1
Thanks Samuli. I think I found the solution, I will reply there on the issue page.
DeleteI also noticed some changes on atom names (e.g. C310 becomes 0C31) when converting gro files to pdb using GROMACS tools. I have written an issue
Deletehttps://github.com/patrickfuchs/autoLipMap/issues/2
@Samuli: could you please comment there?