I have now made some significant changes in the arrangement of the simulation details part of the manuscript: the simulation details texts are now in the Supplementary section and references to the simulation files and details are added to the tables describing the simulated systems. There is also updated ToDo list. For these reasons I have now made a pdf with new file name:
https://www.dropbox.com/s/us0d805lpg53kqe/HGmodel_draft2.pdf?dl=0
You can still access the previous draft version using the links in previous post.
There are also some changes in the content (marked with bold) compared to the previous version.
The goal is to submit the manuscript to the arXiv server latest at 14.1.2015 due to the grant application deadline.
Important note:
I think that it would be very useful if we would share as much simulation files as possible. Since Zenodo allows 2 GB files, also the trajectory sharing is possible. I have created the nmrlipids community in the Zenodo where some simulation files with trajectories related to this project are already shared. The more we get trajectories and other files there the better. To exemplify the potential utility of this collection, I wrote a script which automatically dowloads simulation data from Zenodo and calculates the form factor (FFcomp.sh, see also discussion with Peter Heftberger). Note that Zenodo gives a doi indentification for your data, thus it can be cited. In the current version of the manuscript these citations are used in the simulation system tables to refer the location of files. If someone does not want or cannot share the files, we can just leave these table items empty.
Tuesday, December 23, 2014
Friday, November 21, 2014
New version of the manuscript
First of all, I want to thank all the contributors and readers for their courage to participate to this project. I am impressed about people's willingness to contribute for such a project. I think that we have proven that this approach can be very effective in our field.
Originally one of the ideas of the project was to improve the manuscript that was published in the arXiv. I have now basically rewritten the original manuscript based on the results published in this blog. You can find the current version of the manuscript here: https://www.dropbox.com/s/kza9ta2kpu54jod/HGmodel_forComments.pdf?dl=0
There is also the version which is constantly updated during the writing process found here:
https://www.dropbox.com/s/0xlxndnh1ezik2g/HGmodel_draft.pdf?dl=0
The current manuscript covers only the results for fully hydrated bilayers, effect of dehydration and effect of cholesterol. The plan is to write a separate manuscript about the ion-lipid interactions.
There are still several open issues and missing details in the manuscript (listed in a todo list in the manuscript). Once these have been sorted out, the manuscript will be published in arXiv and also submission to a Journal will be considered.
All kinds of comments about the manuscript are welcomed from everybody (also from people who are not contributors yet). You can submit your comments by commenting this post. The *tex file of the manuscript is also available in GitHub. If you want, you can directly modify that and make a pull request to include your modifications into the manuscript.
There are some very important practical notes:
Authorship. The authorship of the manuscript will be offered for everyone who has commented the blog, according to the "on the credits post". The final decision on authorship will be made by invited individuals themselves based on their self-assessment of their scientific contribution to the project. I have sent the manuscript to the contributors few days a ago asking if their name should be included in the author list. I have currently included the names of authors who have clearly and directly answered to the question "Will your name be included in the authorlist of the current manuscript?" To become an author you have to straightforwardly answer that question through the email or by commenting this post (i.e. sending simulation details etc. is not interpreted as a yes answer to the question about the authorship).
Comments, questions and contributions. One of the key ideas of this project is that all the discussion about the scientifc content will be done publicly through this blog. This implies also to the comments, questions and contributions regarding the manuscript. Since I was asking contributor's decisions about authorship by email, I got some replies related to the content also by email. Due to the idea of having all the discussion about content publicly available, I will repeat here the comments, questions and contributions I have received by email. Ideally further discussion related to the content of the manuscript will continued by commenting this post.
COMMENTS AND QUESTIONS FROM EMAILS:
1) Matti Javanainen commented (translated from Finnish):
Time constants in Gromacs are in ps, not in (ps)^-1.
I would not report real space cut-off's anymore since, at least, in the Gromacs 4.6. and newer they are optimized in the beginning of each simulation, thus the values set in mdp file do not have any practical meaning.
SAMULI: We should check from which version this optimization thing has started.
2) Antti Lamberg asked (translated from Finnish): Why do we have only absolute values in Fig. 2.? In this figure my parameters would be good, however, they have the wrong signs.
SAMULI: The real reason is that the most of the data in Fig. 2 was contributed and the figure was made before we started to look at the signs and stereospecific labeling (largely due to your contributions). We could (and it would be somewhat useful as well) to include also signs and stereospecific labeling into these results. However, it would not change the conclusions made from that figure, i.e. that only CHARMM36, MacRog and GAFFlipid are worth of more detailed studies (now taking into account only published models). For this reason my opinion is that we should not use our time and energy to include all the details into the Fig. 2. My idea is currently that we would leave the attemptions to improve the model for further publications, and I think that in this context we could discuss the issue that there may be a model which has good absolute values, but wrong signs and stereospecific labeling. Since this does not happen in practise with the published models discussed in the current manuscript we can leave this discussion out now.
CONTRIBUTIONS FROM EMAILS:
A lot of information from Matti Javanainen:
https://www.dropbox.com/s/3ru4rbba4a02vm8/commentsFROMmatti.pdf?dl=0
Andrea Catte send the simulation details from his simulations (these contained ions, thus they are not used in the current manusript but will be relevant for the manuscript about ion-lipid interactions):
The starting DPPC lipid bilayer, which was built with the online CHARMM-GUI
(http://www.charmm-gui.org/), contained 600 lipids, 30 water molecules/lipid, Na+ and Cl- ions (150 mM NaCl). The TIP3p water model was used to solvate the system. AA MD simulations of DPPC lipid bilayers were performed at ten different temperatures (283, 298, 303, 308, 312, 313, 314, 318, 323, and 333 K) using the GROMACS software package version 4.5.5 and the S tockholm lipids (S lipids) force field parameters for phospholipids. After energy minimization and a short equilibration run of 50 ps (time step 1fs), 100 ns production runs were performed using a time step of 2 fs with leap-frog integrator. All covalent bonds were constrained with the LINCS algorithm. Coordinates were written every 100 ps. PME with real space cut-off at 1.0 nm was used for Coulomb interactions. Lennard-Jones (LJ) interactions were switched to zero between 1.0 nm and 1.4 nm. The neighbour lists were updated every 10th step with a cut-off of 1.6 nm. Temperature was coupled separately for upper and bottom leaflets of the lipid bilayer, and for water to one of the temperatures reported above with the Nosè-Hoover thermostat using a time constant of 0.5 ps. Pressure was semi-isotropically coupled to the atmospheric pressure with the Parrinello-Rahman barostat using a time constant of 10 ps. The last 40 ns of each simulation was employed for the analysis of DPPC choline and glycerol backbone order parameters.

Originally one of the ideas of the project was to improve the manuscript that was published in the arXiv. I have now basically rewritten the original manuscript based on the results published in this blog. You can find the current version of the manuscript here: https://www.dropbox.com/s/kza9ta2kpu54jod/HGmodel_forComments.pdf?dl=0
There is also the version which is constantly updated during the writing process found here:
https://www.dropbox.com/s/0xlxndnh1ezik2g/HGmodel_draft.pdf?dl=0
The current manuscript covers only the results for fully hydrated bilayers, effect of dehydration and effect of cholesterol. The plan is to write a separate manuscript about the ion-lipid interactions.
There are still several open issues and missing details in the manuscript (listed in a todo list in the manuscript). Once these have been sorted out, the manuscript will be published in arXiv and also submission to a Journal will be considered.
All kinds of comments about the manuscript are welcomed from everybody (also from people who are not contributors yet). You can submit your comments by commenting this post. The *tex file of the manuscript is also available in GitHub. If you want, you can directly modify that and make a pull request to include your modifications into the manuscript.
There are some very important practical notes:
Authorship. The authorship of the manuscript will be offered for everyone who has commented the blog, according to the "on the credits post". The final decision on authorship will be made by invited individuals themselves based on their self-assessment of their scientific contribution to the project. I have sent the manuscript to the contributors few days a ago asking if their name should be included in the author list. I have currently included the names of authors who have clearly and directly answered to the question "Will your name be included in the authorlist of the current manuscript?" To become an author you have to straightforwardly answer that question through the email or by commenting this post (i.e. sending simulation details etc. is not interpreted as a yes answer to the question about the authorship).
Comments, questions and contributions. One of the key ideas of this project is that all the discussion about the scientifc content will be done publicly through this blog. This implies also to the comments, questions and contributions regarding the manuscript. Since I was asking contributor's decisions about authorship by email, I got some replies related to the content also by email. Due to the idea of having all the discussion about content publicly available, I will repeat here the comments, questions and contributions I have received by email. Ideally further discussion related to the content of the manuscript will continued by commenting this post.
COMMENTS AND QUESTIONS FROM EMAILS:
1) Matti Javanainen commented (translated from Finnish):
Time constants in Gromacs are in ps, not in (ps)^-1.
I would not report real space cut-off's anymore since, at least, in the Gromacs 4.6. and newer they are optimized in the beginning of each simulation, thus the values set in mdp file do not have any practical meaning.
SAMULI: We should check from which version this optimization thing has started.
2) Antti Lamberg asked (translated from Finnish): Why do we have only absolute values in Fig. 2.? In this figure my parameters would be good, however, they have the wrong signs.
SAMULI: The real reason is that the most of the data in Fig. 2 was contributed and the figure was made before we started to look at the signs and stereospecific labeling (largely due to your contributions). We could (and it would be somewhat useful as well) to include also signs and stereospecific labeling into these results. However, it would not change the conclusions made from that figure, i.e. that only CHARMM36, MacRog and GAFFlipid are worth of more detailed studies (now taking into account only published models). For this reason my opinion is that we should not use our time and energy to include all the details into the Fig. 2. My idea is currently that we would leave the attemptions to improve the model for further publications, and I think that in this context we could discuss the issue that there may be a model which has good absolute values, but wrong signs and stereospecific labeling. Since this does not happen in practise with the published models discussed in the current manuscript we can leave this discussion out now.
CONTRIBUTIONS FROM EMAILS:
A lot of information from Matti Javanainen:
https://www.dropbox.com/s/3ru4rbba4a02vm8/commentsFROMmatti.pdf?dl=0
Andrea Catte send the simulation details from his simulations (these contained ions, thus they are not used in the current manusript but will be relevant for the manuscript about ion-lipid interactions):
The starting DPPC lipid bilayer, which was built with the online CHARMM-GUI
(http://www.charmm-gui.org/), contained 600 lipids, 30 water molecules/lipid, Na+ and Cl- ions (150 mM NaCl). The TIP3p water model was used to solvate the system. AA MD simulations of DPPC lipid bilayers were performed at ten different temperatures (283, 298, 303, 308, 312, 313, 314, 318, 323, and 333 K) using the GROMACS software package version 4.5.5 and the S tockholm lipids (S lipids) force field parameters for phospholipids. After energy minimization and a short equilibration run of 50 ps (time step 1fs), 100 ns production runs were performed using a time step of 2 fs with leap-frog integrator. All covalent bonds were constrained with the LINCS algorithm. Coordinates were written every 100 ps. PME with real space cut-off at 1.0 nm was used for Coulomb interactions. Lennard-Jones (LJ) interactions were switched to zero between 1.0 nm and 1.4 nm. The neighbour lists were updated every 10th step with a cut-off of 1.6 nm. Temperature was coupled separately for upper and bottom leaflets of the lipid bilayer, and for water to one of the temperatures reported above with the Nosè-Hoover thermostat using a time constant of 0.5 ps. Pressure was semi-isotropically coupled to the atmospheric pressure with the Parrinello-Rahman barostat using a time constant of 10 ps. The last 40 ns of each simulation was employed for the analysis of DPPC choline and glycerol backbone order parameters.
Alexander Lyubartsev sent some simulation details:
Here are details of simulations with Hörberg et al force field:
lipid Nl Nw T(K) t-sim t-an
DMPC 98 2700 303 75 50
POPC 128 3840 303 100 80
DMPC simulations are from ref 19 (Högberg et al)
POPC simulations are from A.L.Rabinovich, A.P.Lyubartsev J.Phys.
Conference series
510 , 012022 (2014)
Simulation details:
Time step 2fs, Ewald summation,
cut-off 14 Å, update of neighbour list each 10 steps,
Nose-Hoover thermostat, Parrinello-Rahman barostat,
semi-anizotropic pressure coupling,
long-range isotropic pressure correction
Final note by Samuli: This suggestion in Page 7 in the manuscript:
"Interestingly, the probability of the gaughe
conformations correlates with the order parameter difference
between the \(\beta\) and \(\alpha\) segments: the larger the gaughe fraction
the larger the order parameter difference. This suggestion together
with the results in Fig. 3 would indicate that the correct
gaughe-trans fraction for N- - -O dihedral is larger than in
GAFFlipid but smaller than in CHARMM36."
is not supported by the recent results in the blog from the new model by
Tjörnhammar and Edholm. I will rewrite this discussion.
lipid Nl Nw T(K) t-sim t-an
DMPC 98 2700 303 75 50
POPC 128 3840 303 100 80
DMPC simulations are from ref 19 (Högberg et al)
POPC simulations are from A.L.Rabinovich, A.P.Lyubartsev J.Phys.
Conference series
510 , 012022 (2014)
Simulation details:
Time step 2fs, Ewald summation,
cut-off 14 Å, update of neighbour list each 10 steps,
Nose-Hoover thermostat, Parrinello-Rahman barostat,
semi-anizotropic pressure coupling,
long-range isotropic pressure correction
Final note by Samuli: This suggestion in Page 7 in the manuscript:
"Interestingly, the probability of the gaughe
conformations correlates with the order parameter difference
between the \(\beta\) and \(\alpha\) segments: the larger the gaughe fraction
the larger the order parameter difference. This suggestion together
with the results in Fig. 3 would indicate that the correct
gaughe-trans fraction for N- - -O dihedral is larger than in
GAFFlipid but smaller than in CHARMM36."
is not supported by the recent results in the blog from the new model by
Tjörnhammar and Edholm. I will rewrite this discussion.
Monday, September 1, 2014
About glycerol conformations
[Update 12.11.2014] Originally this post claimed that the GAFFlipid model has the wrong isomer for the glycerol backbone. Further studies revealed that the problem was actually the initial configuration downloaded from http://lipidbook.bioch.ox.ac.uk/package/show/id/150.html having different isomers in different leaflets (see comments to this post). After setting up new simulations with correct initial stucture also the GAFFlipid has the correct stereospecifity for the g\(_1\) segment:
[Original post begins here:]
Based on discussions in the blog and the experiments by Gally et al. we have now reported the order parameter results by taking the stereospecifity of g\(_1\) and g\(_3\) into account. The result was already reported in the Current status of the project on 29.4.2014, and it is shown here again in Fig. 1.
 |
| Figure 1. Order parameters for choline and glycerol for POPC. Magnitudes measured by Ferreira et al., signs measured by Hong et al., Hong et al. and Gross et al. , and stereospecific labeling measured by Gally et al. |
[Original post begins here:]
Based on discussions in the blog and the experiments by Gally et al. we have now reported the order parameter results by taking the stereospecifity of g\(_1\) and g\(_3\) into account. The result was already reported in the Current status of the project on 29.4.2014, and it is shown here again in Fig. 1.
 |
| Figure 1. Order parameters for choline and glycerol for POPC. Magnitudes measured by Ferreira et al., signs measured by Hong et al., Hong et al. and Gross et al. , and stereospecific labeling measured by Gally et al. |
The main conclusion from the Fig. 1 was that the GAFFlipid model has different stereospecifity in g\(_1\) order parameters compared to CHARMM36, MacRog and experiments. While writing the new version of manuscript I was plotting the lipid structures from different simulations and noticed that the GAFFlipid has different isomer in glycerol compared to other models. This is demonstrated in Fig. 2
 |
| Figure 2. Snapshots of lipid structures from simulations with different models. All the structures are shown such that the viewer is looking at the direction of g\(_1\)-g\(2\) bond. In Berger, MacRog and CHARMM36 the g\(_2\) hydrogen is pointing to the right. In GAFFlipid it is pointing left. |
In conclusion, the different isomer in GAFFlipid explains the different R/S labeling observed in simulations. I did also check the isomer in Lipid14 and it seems that the issue has been fixed there (Lipid14 had the magnitudes more off from experiments, however, as seen in the The lipid forcefield comparison post).
Wednesday, August 20, 2014
Presentations in International Workshop on Biomembranes — From Fundamentals to Applications
International Workshop on Biomembranes — From Fundamentals to Applications was organized 19.8.-22.8.2014 at CSC-IT Center for Science, Finland.
Markus Miettinen gave a talk and Samuli Ollila presented a poster describing the nmrlipids project. The presentations can be found below. If you have any questions or comments about the presentations you are welcome to comment this post. You can do this whether you were present in the meeting or not.
Markus Miettinen gave a talk and Samuli Ollila presented a poster describing the nmrlipids project. The presentations can be found below. If you have any questions or comments about the presentations you are welcome to comment this post. You can do this whether you were present in the meeting or not.
Talk (slides with no audio) presented by Markus Miettinen.
 |
| Poster presented by Samuli Ollila. |
Monday, May 19, 2014
Towards a new version of the manuscript
In the opening post of the nmrlipids project, published simultaneously with the accompanying manuscript, it was written that:
With these refinements (sign and R/S labeling) it becomes clear that the order parameters for the g\(_1\) group in the GAFFlipid force field are not in a reasonable agreement with experiments. Thus there are only two all-atom models left to consider: CHARMM36 and MacRog. Interestingly, as discussed in the post Response of headgroup and glycerol order parameters to changing conditions: Results, the responses of these two force fields to changing conditions (dehydration, ion concentration and cholesterol content) are in qualitative agreement with experiments. However, the Na\(^+\) association with the bilayer is clearly too strong for MacRog; the association is less for CHARMM36, but more simulations are needed to complete the comparison to experiments.
In conclusion, CHARMM36 is a force field that relatively well satisfies the original goal of the nmrlipids project. In this respect, we could now just write up the manuscript, submit and finish the project.
Having said that, I still think that it would be very useful to have also a united-atom model that fulfills the original goal. Regarding the studies of pure lipid bilayers, CHARMM36 appears significantly slower than united-atom models and also slower than other all-atom models. Based on my personal experience (I have not investigated it further) I think the slowness is due to the Lennard-Jones interactions of the water hydrogens in the CHARMM water model\(^2\). In addition to speed, there will probably be interest in using a lipid model with correct molecular structure in combination with proteins (or other molecules) that lack a model combatible with the CHARMM36 lipids.
If we manage to get correct structures for one united atom model, the same procedure could probably be used for other models as well. A promising method to tune the potentials to get the correct order parameters was already reported in this blog by Antti Lamberg in March. However, at the time, we had not considered the signs and the R/S labeling and thus the order parameters were matched to wrong values.
On the basis of these perpectives, I suggest that we make one further attempt to correct the order parameters for the Berger model in the following way:
First results for step 1 of the procedure. As preliminary work for the above procedure I plotted 40 consecutive lipid structures observed in Berger and CHARMM36 simulations such that the glycerol groups were aligned. The results are shown in Figs. 2 and 3.
Comparing the Figs. 2 and 3 one can clearly see that the conformations sampled by the g\(_1\) and g\(_3\) groups significantly differ between the models - which is no surprise as the models predict different order parameters. In all configurations of the CHARMM36 model the hydrogens of the g\(_1\) group are pointing to the same direction, indicating a rather rigid dihedral in the g\(_1\)-g\(_2\) bond. In the Berger model, the g\(_1\)-g\(_2\) bond seems to be significantly more flexible. The situation is opposite for the g\(_3\) group: the hydrogens are pointing in the same direction in almost all configurations for the Berger model, while CHARMM36 seems to be significantly more flexible. This difference seems to have significant effect to the conformations of the whole PC headgroup. (When looking at the figures, it should be noted that the molecules are shown from different angles to make the above points more clear.)
I am currently processing some ideas how to proceed to the step 2 in the procedure. I will report the progress in the near future. It would be also interesting to see structural figures with aligned glycerol from the MacRog model.
\(^1\)The recent united atom CHARMM has not been tested yet. The authors of the model state that "The headgroup is still an AA potential in C36-UA, so only the chain SCDs were compared." Thus similar results than in CHARMM36 are expected. However, the united atom CHARMM might still be quite slow due to the water model. Some results with the model would be interesting.
\(^2\)Using regular TIP3P model does not seem to be a solution since significant changes in phase behaviour was reported by Piggot et al.
"The ultimate goal of this blog is to find an atomistic (preferably united-atom) force field that reproduces the experimental properties discussed in the manuscript. Naturally the optimal situation would be that some of the already available force fields would fulfill this goal. If this, however, turns out not to be the case, the goal will be to find the appropriate modifications."As discussed in The lipid forcefield comparison post, none of the tested united-atom force fields fulfills the goal\(^1\). The same post concluded that three all-atom force fields (CHARMM36, GAFFlipids and MacRog) gave a reasonable agreement with experiments for a fully hydrated pure bilayer, when the absolute values of headgroup and glycerol order parameters were compared. Since then, the comparison has been refined to include the sign of the order parameters as well as the R/S labeling of hydrogens. Fig. 1 shows an up to date comparison of the top three all-atom force fields, the Berger force field, and the Lamberg parameters (currently in development in this blog) to experiments.
 |
| Figure 1. A set of simulation results for the POPC headgroup and glycerol order parameters reported in the blog is compared to experiments. |
In conclusion, CHARMM36 is a force field that relatively well satisfies the original goal of the nmrlipids project. In this respect, we could now just write up the manuscript, submit and finish the project.
Having said that, I still think that it would be very useful to have also a united-atom model that fulfills the original goal. Regarding the studies of pure lipid bilayers, CHARMM36 appears significantly slower than united-atom models and also slower than other all-atom models. Based on my personal experience (I have not investigated it further) I think the slowness is due to the Lennard-Jones interactions of the water hydrogens in the CHARMM water model\(^2\). In addition to speed, there will probably be interest in using a lipid model with correct molecular structure in combination with proteins (or other molecules) that lack a model combatible with the CHARMM36 lipids.
If we manage to get correct structures for one united atom model, the same procedure could probably be used for other models as well. A promising method to tune the potentials to get the correct order parameters was already reported in this blog by Antti Lamberg in March. However, at the time, we had not considered the signs and the R/S labeling and thus the order parameters were matched to wrong values.
On the basis of these perpectives, I suggest that we make one further attempt to correct the order parameters for the Berger model in the following way:
- We calculate the dihedral distributions from the CHARMM36 (and MacRog) for the glycerol and headgroup region.
- We set up dihedral potentials for the Berger model such that they resemble the ones in the CHARMM36 (and/or MacRog).
- We use these potentials as a starting point for a Lamberg-style optimization.
First results for step 1 of the procedure. As preliminary work for the above procedure I plotted 40 consecutive lipid structures observed in Berger and CHARMM36 simulations such that the glycerol groups were aligned. The results are shown in Figs. 2 and 3.
 |
| Figure 2. 40 consecutive (\(\Delta\)t=1.25 ns) structures observed in a Berger simulations (POPC, T=298K). Hydrogens have been added to the visualization by the Gromacs protonate program in the same way as for the order parameter calculation. |
 |
| Figure 3. 40 consecutive (\(\Delta\)t=1.25 ns) structures observed in a CHARMM36 simulation (POPC, T=303K). |
I am currently processing some ideas how to proceed to the step 2 in the procedure. I will report the progress in the near future. It would be also interesting to see structural figures with aligned glycerol from the MacRog model.
\(^1\)The recent united atom CHARMM has not been tested yet. The authors of the model state that "The headgroup is still an AA potential in C36-UA, so only the chain SCDs were compared." Thus similar results than in CHARMM36 are expected. However, the united atom CHARMM might still be quite slow due to the water model. Some results with the model would be interesting.
\(^2\)Using regular TIP3P model does not seem to be a solution since significant changes in phase behaviour was reported by Piggot et al.
Friday, May 2, 2014
Response of headgroup and glycerol order parameters to changing conditions: Results
During our project, the responses of headgroup and glycerol order parameters to hydration level, ion concentration and cholesterol content have already been reported for several models, and the results have been discussed to some extent in various comments. This post reviews the results for the responses reported in the nmrlipids-project so far.
Note on signs.
In the original manuscript and in the plots previously published in the blog (dehydration, NaCl, CaCl), the responses of the order parameters were discussed only in terms of their absolute values. From these plots the response of the \(\beta\)-carbon order parameter in all force fields except in CHARMM seemed to be qualitatively different than in experiments. Recently we have started to consider also the signs of the order parameters in addition to their magnitude. Even though the signs are not explicitly measured when responses are considered experimentally, it is reasonable to assume that the signs are not changing except for the case of adding a large amount of charges to the bilayer. Throughout this posting the signs of the order parameters are taken into account. For dehydration and ions the signs are relevant for the conclusions
The order parameters as a function of hydration level. It is clear from Fig. 1 that in experiments dehydration leads to more positive order parameters for the \(\beta\) and \(\alpha\) carbons. Results from all the experiments are in general very similar, despite some variation in lipid compositions and temperatures. The glycerol order parameters have been measured only in one experiment, where a decrease with dehydration was observed for the g\(_3\) and g\(_2\), while g\(_1\) remains unchanged.
In simulations, as in experiments, the \(\beta\)- and \(\alpha\)-carbon order parameters increase with dehydration - even though the magnitudes are significantly different between different models and some models show significant forking. Note that this qualitative agreement between simulations and experiments for the \(\beta\)-carbon dehydration response differs from the conclusions of the original manuscript and of the discussions in the nmrlipids-blog. The reason for this is that previously only the absolute values have been discussed and many many force fields give wrong sign for the \(\beta\)-carbon order parameter: Increase in a negative order parameter decreases its absolute value (as seen in the experiments) while increase in a positive order parameter increases its absolute value (as seen in most simulations).
When the comparison between simulations and experiments is extended to the glycerol region, only the CHARMM36 force field can be interpreted to have all the order parameters quantitatively close to experiments, and to have qualitatively correct response to dehydration. However, the changes in the order parameters and difference between MacRog results and experiments are relatively small in the glycerol region.
The order parameters as a function of NaCl or CaCl\(_2\) concentration. Fig. 2 shows the effect of NaCl on headgroup and glycerol order parameters.
The effect of penetrating charges on lipid bilayer properties has often been studied with NMR by measuring the \(\beta\) and \(\alpha\) order parameters [Seelig et al. (1987), Semchyschyn et al. (2004) and references therein]. For this reason let us take a closer look of these order parameters as a function of NaCl and CaCl\(_2\) (Fig. 3).
It is clear from Figs. 2 and 3 that NaCl induces a dramatic decrease of the \(\beta\)- and \(\alpha\)-carbon order parameters in Berger and MacRog models; the effect is too strong compared to the experiments. Qualitatively, however, this does correspond to the experimentally observed effect of penetrating charges (multivalent ions, anionic surfactants) into PC bilayers [Seelig et al. (1987)]. This qualitative correspondence was hidden in our previous considerations (NaCl, CaCl) in which only the absolute values were compared (similarly to the dehydration case, discussed above). The most obvious explanation for this finding is that the Na\(^+\)-partitioning into the bilayer is too strong in Berger and MacRog models.
The situation is different in CHARMM36 and Orange force fields: Adding NaCl induces small or nonexistent changes at the \(\beta\) and \(\alpha\) order parameters. This signals significantly lower partitioning. The data as a function of CaCl\(_2\) with Orange force field (Fig. 4), however, shows a disctinct difference to the Na\(^+\) case: Ca\(^{2+}\) significantly reduces the \(\beta\)- and \(\alpha\)-order parameters. This disctinction has been seen also in experiments, where it has been interpreted as a signature of the different in partitioning of Na\(^{+}\) and Ca\(^{2+}\) into PC bilayers. (Note, however, that in the Orange force field addition of Ca\(^{2+}\) leads to changes in order parameters that are larger than in experiments.)
To check the validity of interpreting the order parameter results in terms of differences partitioning affinities, Fig. 4 shows the density profiles for lipids and ions in different simulations.
By comparing Figs 2, 3 and 4 it becomes clear that Na\(^+\) binding is stronger (Fig. 4) in the force fields in which the Na\(^+\)-induced changes in the order parameters are larger (Figs. 2 and 3), i.e., in Berger and MacRog. In CHARMM36 and Orange, in which the Na\(^+\)-induced changes in order parameters are smaller, also the partitioning of Na\(^+\) is significantly less. It seems that the correlation between ion binding and order parameter changes that was suggested from the NMR experiments [Akutsu et al. (1981), Seelig et al. (1987)] can be reproduced in MD simulations. This is an important finding. However, to conclude how straightforward the connection between order parameters and ion partitioning is, and if the binding affinity in the Orange and CHARMM36 force fields is in agreement with experiments, we have to make more quantitative analysis on binding and run simulations with different ion concentrations.
As a final note in ions, it is interesting that the Berger and Orange simulations used the same ion model. Thus, the Na\(^+\) partitioning can be largely reduced just by modifying the lipid parameters. More detailed analysis of the relevant changes, however, is difficult since the Orange parameters are not yet publicly available.
The order parameters as function of cholesterol concentration. Based on the reported data, both force fields (MacRog and CHARMM36) reproduce the response to cholesterol relatively well (Fig. 5). This was not the case for the Berger, as discussed in the original manuscript. However, for MacRog high cholesterol contents have not been reported and it is still possible for discrepancies to occur there.
Preliminary conclusions
Note on signs.
In the original manuscript and in the plots previously published in the blog (dehydration, NaCl, CaCl), the responses of the order parameters were discussed only in terms of their absolute values. From these plots the response of the \(\beta\)-carbon order parameter in all force fields except in CHARMM seemed to be qualitatively different than in experiments. Recently we have started to consider also the signs of the order parameters in addition to their magnitude. Even though the signs are not explicitly measured when responses are considered experimentally, it is reasonable to assume that the signs are not changing except for the case of adding a large amount of charges to the bilayer. Throughout this posting the signs of the order parameters are taken into account. For dehydration and ions the signs are relevant for the conclusions
The order parameters as a function of hydration level. It is clear from Fig. 1 that in experiments dehydration leads to more positive order parameters for the \(\beta\) and \(\alpha\) carbons. Results from all the experiments are in general very similar, despite some variation in lipid compositions and temperatures. The glycerol order parameters have been measured only in one experiment, where a decrease with dehydration was observed for the g\(_3\) and g\(_2\), while g\(_1\) remains unchanged.
 |
| Figure 1. Order parameters as a function of hydration level from the simulations reported through this blog and from the experiments by Dvinskikh et al. (2005), Ulrich et al. (1994) and Bechinger et al. (1991) In these experiments only absolute values were measured. The signs are assumed to be the same as in fully hydrated conditions measured by Hong et al. (1995), Hong et al. (1995) and Gross et al. (1997). Simulation values for some carbon segments are not seen since they are beyond the scale of the y-axis. |
In simulations, as in experiments, the \(\beta\)- and \(\alpha\)-carbon order parameters increase with dehydration - even though the magnitudes are significantly different between different models and some models show significant forking. Note that this qualitative agreement between simulations and experiments for the \(\beta\)-carbon dehydration response differs from the conclusions of the original manuscript and of the discussions in the nmrlipids-blog. The reason for this is that previously only the absolute values have been discussed and many many force fields give wrong sign for the \(\beta\)-carbon order parameter: Increase in a negative order parameter decreases its absolute value (as seen in the experiments) while increase in a positive order parameter increases its absolute value (as seen in most simulations).
When the comparison between simulations and experiments is extended to the glycerol region, only the CHARMM36 force field can be interpreted to have all the order parameters quantitatively close to experiments, and to have qualitatively correct response to dehydration. However, the changes in the order parameters and difference between MacRog results and experiments are relatively small in the glycerol region.
The order parameters as a function of NaCl or CaCl\(_2\) concentration. Fig. 2 shows the effect of NaCl on headgroup and glycerol order parameters.
 |
| Figure 2. Order parameters as a function of NaCl concentration from simulations reported in the blog and from experiments by Akutsu et al. (1981). The signs are assumed to be the same as measured by Hong et al. (1995), Hong et al. (1995) and Gross et al. (1997). The effect of ions to the g\(_2\) and g\(_3\) were not measured, thus only the values without ions are shown. The straight line between the results with and without ions is plotted to guide the eye. The results with ions for the GAFFlipids are not available. The results for the Lamberg model are not included since the model is still under development. |
The effect of penetrating charges on lipid bilayer properties has often been studied with NMR by measuring the \(\beta\) and \(\alpha\) order parameters [Seelig et al. (1987), Semchyschyn et al. (2004) and references therein]. For this reason let us take a closer look of these order parameters as a function of NaCl and CaCl\(_2\) (Fig. 3).
 |
| Figure 3. Order parameters for \(\beta\) and \(\alpha\) carbons as a function of NaCl (left column) and CaCl\(_2\) (right column) concentrations from simulations reported in the blog and from the experiments by Akutsu et al. (1981). The Berger results are shown in the plot even though some of their values are beyond the y-axis scale. |
The situation is different in CHARMM36 and Orange force fields: Adding NaCl induces small or nonexistent changes at the \(\beta\) and \(\alpha\) order parameters. This signals significantly lower partitioning. The data as a function of CaCl\(_2\) with Orange force field (Fig. 4), however, shows a disctinct difference to the Na\(^+\) case: Ca\(^{2+}\) significantly reduces the \(\beta\)- and \(\alpha\)-order parameters. This disctinction has been seen also in experiments, where it has been interpreted as a signature of the different in partitioning of Na\(^{+}\) and Ca\(^{2+}\) into PC bilayers. (Note, however, that in the Orange force field addition of Ca\(^{2+}\) leads to changes in order parameters that are larger than in experiments.)
To check the validity of interpreting the order parameter results in terms of differences partitioning affinities, Fig. 4 shows the density profiles for lipids and ions in different simulations.
 |
| Figure 4. Density profiles for lipids and ions from different simulations reported in the nmrlipids-blog. The ion densitied are multiplied with 4 in MacRog, and with 50 in other models to make them visible with the used y-axis scale. |
As a final note in ions, it is interesting that the Berger and Orange simulations used the same ion model. Thus, the Na\(^+\) partitioning can be largely reduced just by modifying the lipid parameters. More detailed analysis of the relevant changes, however, is difficult since the Orange parameters are not yet publicly available.
The order parameters as function of cholesterol concentration. Based on the reported data, both force fields (MacRog and CHARMM36) reproduce the response to cholesterol relatively well (Fig. 5). This was not the case for the Berger, as discussed in the original manuscript. However, for MacRog high cholesterol contents have not been reported and it is still possible for discrepancies to occur there.
 |
| Figure 5. Order parameters as a function of cholesterol concentration from simulations reported in the blog and from experiments by Ferreira et al. (signs taken from Hong et al. (1995), Hong et al. (1995) and Gross et al. (1997) and assumed to be unchanged with cholesterol concentration). In the CHARMM36 results the cholesterol force field by Lim et al. (2012) was used.The results from Berger force field are shown even though some values are beyond the y-axis scale. For other force fields the data is not available. |
- When the signs are taken into account, the qualitative response to dehydration and penetrating charges seems to agree with experiments in all tested force fields.
- As suggested in experiments, the changes in headgroup order parameters can be related to the amount on penetrated charge also in simulations.
- Comparison to experiments shows Na\(^+\) partitioning to be too strong in Berger and MacRog models.
- More detailed studies are needed to conclude if Na\(^+\) partitioning in CHARMM36 and Orange agrees with experiments.
- Response to cholesterol seems to be relative realistic in CHARMM36 and MacRog.
Thursday, April 10, 2014
On the signs of the order parameters
[updated 7.5.2014]
Until now, only the absolute values of order parameters have been looked at in the nmrlipids project. However, already the first comment to this blog pointed out the relevance of the signs. (I was a little bit sloppy in my reply and said that the signs of the order parameters are correct in Berger; this is not true as mentioned here and dicussed below). It is important to take the sign into account when the order parameters are discussed in relation to the structure of lipid molecules: Two C-H bonds whose order parameters have the same magnitude but different signs are sampling different orientations. Consequently, the correct signs are crucial if force field parameters are fitted to reproduce the correct order parameters, for example, by using the method suggested by Antti Lamberg in this blog. Thus, I think that from now on we have to start looking also at the signs of the order parameters, not just their magnitude.
Until now, only the absolute values of order parameters have been looked at in the nmrlipids project. However, already the first comment to this blog pointed out the relevance of the signs. (I was a little bit sloppy in my reply and said that the signs of the order parameters are correct in Berger; this is not true as mentioned here and dicussed below). It is important to take the sign into account when the order parameters are discussed in relation to the structure of lipid molecules: Two C-H bonds whose order parameters have the same magnitude but different signs are sampling different orientations. Consequently, the correct signs are crucial if force field parameters are fitted to reproduce the correct order parameters, for example, by using the method suggested by Antti Lamberg in this blog. Thus, I think that from now on we have to start looking also at the signs of the order parameters, not just their magnitude.
Note that in the extensive comparison of the headgroup and glycerol order parameters in fully hydrated lipid bilayers we have gathered for the nmrlipids project only the magnitudes are discussed. I think that (for this project) we do not need to reconsider all these results with the sign, since if the absolute value is not in good agreement with experiment, then the value with sign will not be either.
The signs in experiments: tail region
It can be easily seen from the definition of the C-H order parameter
$$S_{CH}=\frac{3}{2}\langle \cos^2 \theta \rangle - \frac{1}{2}$$
that the possible values are \(-\frac{1}{2} < S_{CH} < +1\). More specifically
$$-\frac{1}{2} < S_{CH} < 0 \quad \mathrm{, when }\quad 0<\langle\cos^2 \theta\ \rangle < \frac{1}{3},$$
and
$$0 < S_{CH} < 1 \quad \mathrm{, when }\quad \frac{1}{3} < \langle\cos^2 \theta\ \rangle < 1.$$
In older publications that used \(^2\)H NMR measurements, only the absolute values of the order parameters (or quadrupolar splittings) were discussed since the sign was not measurable. For the tail C-H vectors, however, the sign was believed to be negative because \(\theta\) was expected to fluctuate around 90\(^{o}\) leading to negative order parameters [Seelig (1977)]. This was later confirmed by using \(^{13}\)C NMR measurements [Hong et al. (1995)]. In simulations, the acyl chain order parameters are typically in good agreement with experiments, including the negative sign (see e.g. Ollila et al (2007)).
The signs in experiments: headgroup and glycerol region
The signs of the ¹³C-H order parameters have been measured by at least two different techniques and groups: Hong et al. first measured eggPC [Hong et al. (1995)] and then DMPC [Hong et al. (1995)]; later Gross et al. used a different NMR technique to measure DMPC [Gross et al. (1997)]. These experiments are in good agreement with one another and report negative order parameters for almost all the segments, only \(\alpha\) and \(\gamma\) are positive.
The signs of the order parameters are also listed in Table 2 of the paper by Aussenac et al. (2003), who conducted ²H NMR measurements for bicelles containing DMPC. However, the signs in the table differ from those reported by Hong et al. (1995), Hong et al. (1995) and Gross et al. (1997): Aussenac et al. give positive signs for one of the hydrogens in g1, g3 and C2 in the acyl chain. This was also noted by Högberg et al. when comparing the results to MD simulations. (The paper by Aussenac et al. is slightly confusing to me since I cannot find any discussion concerning the signs or any description of how they were measured; I would warmly welcome some help from an NMR expert to understand how the signs in the paper by Aussenac have been deduced.)
Update: Patrick Fuchs pointed out that Aussenac et al. have not actually measured the signs but used a model to extract the signs. This confirms the original conclusion that the signs from Hong et al. (1995) and Gross et al. (1997) should be used.
In conclusion, it seems reasonable to assume that the signs measured with ¹³C NMR methods by Hong et al. (1995) and Gross et al. (1997) are correct. Furthermore, it seems that the signs [Hong et al. (1995), Hong et al. (1995), Gross et al. (1997)] and the absolute values [Gally et al. (1981), Ferreira et al. (2013)] are practically unaffected by the acyl tail contents of the bilayers. Thus, I will also assume that the signs for DPPC and POPC bilayers are the same as measured for eggPC and DMPC. To sum up: the signs for almost all the carbons are negative, only \(\alpha\) and \(\gamma\) are positive.
The signs in experiments: varying conditions
Typically when the response of order parameters to varying conditions (ions, dehydration and cholesterol) is measured, only the absolute values are reported. Where clear responses are observed, like for multivalent ions and dehydration, the experiments are done by gradually changing the conditions. The responses of the magnitudes to these changes are systematic, see Figs. 1-3 in the Accuracy post. Thus, it is reasonable to assume that also the signs are not suddenly changing. However, it seems that the sign of the \(\alpha\) carbon order parameter does change in response to a large amount of bound charge, such as multivalent ions. In this case, the absolute value of the order parameter first decreases to zero and then starts to increase again [Altenbach et al. (1984), Seelig et al. (1987)]; the behaviour of the spectra has been nicely illustrated by Altenbach et al., see Fig. 1.
 |
| Figure 1. Quadrupolar splitting \(\Delta \nu_Q\) as a function of CaCl\(_2\) concentration. As discussed in the Accuracy post, this is related to the order parameter as \(S_{CD}=0.00784 \times \Delta \nu_Q\). We know nowadays that the order parameter of \(\alpha\) in the absense of CaCl\(_2\) is positive [Hong et al. (1995), Hong et al. (1995) and Gross et al. (1997)]. Thus, the most obvious interpertation for the result is that the \(\alpha\) order parameter decreases to zero when CaCl\(_2\) concentration reaches 2.0M, and above these concentrations becomes increasingly negative with further addition of CaCl\(_2\). Reprinted with permission from Altenbach and Seelig, Biochemistry, 23, 3913 (1984). Copyright 1984 American Chemical Society. |
Comparison to simulations of fully hydrated pure PC bilayers
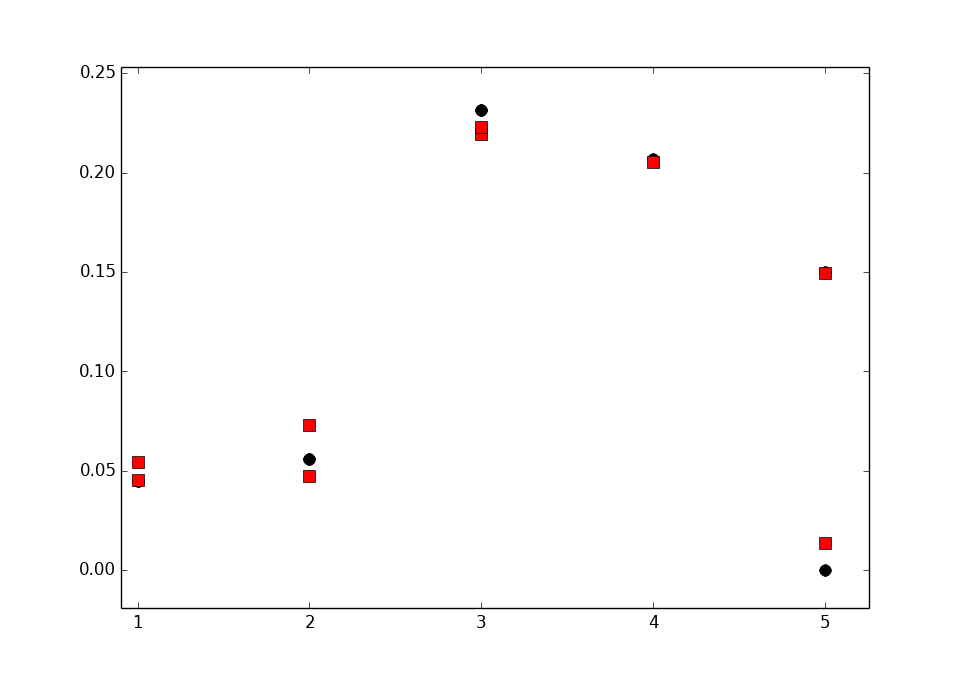
The measured order parameters for the fully hydrated POPC bilayer from Ferreira et al. (2013) with the signs determined by Hong et al. (1995) and Gross et al. (1997) are shown in Fig 2 together with the simulation results for the top 3 force fields (CHARMM36, GAFFlipids and MacRog, see the lipid force field comparison post), the Berger force field, and the modified Berger force field by Antti Lamberg.
 |
| Figure 2. Order parameters with signs from experiments and simulations. Note that the y-axis is inverted to make the figure more easily comparable with the plots with the absolute value of order parameter. The magnitudes for the experimental results are taken from Ferreira et al. (2013) and the signs from Hong et al. (1995) , Hong et al. (1995) and Gross et al. (1997) |
The signs from all the
simulations in Fig. 2 are correct for the carbons \(\alpha\), g3 and
g2. The signs for \(\beta\), however, are positive in simulations with
MacRog, Berger and Lamberg models while experiments give negative
values. In the Berger and Lamberg models also the sign of the larger order
parameter for g1 is positive while negative value is measured in
experiments. This means that the order parameter for the \(\beta\)
carbon in the MacRog model is not as good as it seems when looking
only at absolute values. The same applies for the \(\beta\) and g1 carbons in
the Berger and Lamberg models.
Friday, March 14, 2014
The lipid forcefield comparison
[Updated Friday, April 11, 2014]
Our open research collaboration has tested the abilities of10 12 current force fields to correctly sample the headgroup and glycerol conformations in phospholipids. The C-H order parameters calculated from simulations were compared to the corresponding ones measured in accurate NMR experiments. We found that none of the force fields is perfect, but promising results were obtained with the CHARMM36 and the unpublished Maciejewski–Rog force fields.
Our open research collaboration has tested the abilities of
 |
| Figure 1. The Lipid forcefield comparison. The absolute values of PC-lipid headgroup and glycerol C-H order parameters compared to values measured in NMR experiments. Pure systems, full hydration. The following The computational values have been posted to the nmrlipids.blogspot.fi-blog by the project participants, except for the GAFFlipid and one of the CHARMM36 data sets, which were taken from the original publications [Dickson 2012, Klauda 2010]. The experimental values were taken from the following publications: DMPC 303K from Gross 1997, DMPC 314K from Dvinskikh 2005, DPPC 310K from Gally 1981, DPPC 322K from Seelig 1977, DPPC 323K from Akutsu 1981, and POPC 298K from Ferreira 2013. The vertical bars shown for some of the computational values are not error bars, but demonstrate that for these systems we had two data sets; the ends of the bars mark the values from the two sets, and the dot marks their measurement-time-weighted average. |
Our results are summarised in Fig. 1; in the following we discuss the key points.
Experimental data fall close to one another, irrespective of the lipid tails, temperature, salt (NaCl) concentration, or the experimental technique used. This excellent repeatability has already been discussed at length in Samuli's post on accuracy. Motivated by the high experimental repeatability, we have highlighted in Fig. 1 subjective sweet spots (light blue areas), within which we expect the calculated order parameters of a well-performing force field to fall.
Forking of the order parameters was observed in some cases. (Note that to avoid confusion with the established NMR term splitting—used e.g. to refer to the dipolar splitting in NMR spectra—we use here the word forking to describe the situation where the order parameters of different hydrogens in a single carbon are not equal.)
If the corresponding experimental value is not forked (as is the case for the \(\alpha\)- and \(\beta\)-carbons), forking in the calculated value signals that the simulation is sampling an unrealistic conformation, for which the C-H bond distributions for the hydrogens disagree; this must be considered as a serious shortcoming of the force field. Note, however, that for the g3- and g1-carbons the experimental values do fork (although the forking of the g3 carbon is small and some experiments only report the larger or the average value).
If the corresponding experimental value is not forked (as is the case for the \(\alpha\)- and \(\beta\)-carbons), forking in the calculated value signals that the simulation is sampling an unrealistic conformation, for which the C-H bond distributions for the hydrogens disagree; this must be considered as a serious shortcoming of the force field. Note, however, that for the g3- and g1-carbons the experimental values do fork (although the forking of the g3 carbon is small and some experiments only report the larger or the average value).
None of the forcefields was perfect—three all-atom force fields, however, came close (Fig. 2 focuses on just these three to help comparison):
1) CHARMM36 was the only force field that clearly had no forking in the \(\alpha\)- and \(\beta\)-carbons, but it suffered from slightly too ordered \(\beta\)-, g3- and g1-carbons.
2) Maciejewski–Rog had almost all the values within the sweet spots, but suffered from forking in \(\beta\) (and possibly in \(\alpha\)) as well as a too wide split in g3.
3) GAFFlipid appears generally promising. It suffers, however, from slight forking in \(\alpha\) and \(\beta\), and from too ordered g2 and g1. In addition, we have not been able to reproduce these order parameters so far (see the note on technical issues in the end of this post), and thus show here only the originally reported values [Dickson 2012].
 |
| Figure 2. The same data as in Fig. 1, but only for the three best-performing force fields. |
We note that also the all-atom HoNiLy force field performed quite well in our test, although it did suffer from forking in the \(\alpha\)-carbon, too ordered \(\alpha\)- and g3-carbons, and a too wide forking in g3 (Fig. 1). However, because HoNiLy is a modified CHARMM force field and, in fact, an early development version of the Slipids force field, it appears not urgent to study its further properties.
We also note that by far the best order parameters for the fully hydrated case were reported on March 9th 2014 by Antti Lamberg, who used simple brute force heuristics to optimise the dihedral parameters (of a united atom force field) to accurately reproduce the experimental values. As this approach is very fresh, and currently under discussion, we have not yet included these results into the comparison.
{kind=link}
Conclusion: Next we need the responses. There clearly is still room for improvement in the current force fields, even in the standard system of a pure lipid bilayer at full hydration. However, the quality of a force field (in terms of the goal we set for this project, that is, correctly reproducing the headgroup conformations) depends ultimately on its ability to correctly respond to changing environmental conditions.
Based on the here presented analysis at full hydration, it is now justified to focus mostly on testing the responses—to hydration levels, ion concentrations, and cholesterol content—in the top three force fields: CHARMM36, MacRog, and GAFFlipid. (In fact, some response data have already been reported in the blog. An overview of these will be presented in a separate post.) The responses of the other
Should any of the three promising all-atom force fields pass the response tests, we might try to use it as a basis on which to build a well-performing united atom model. If successful, this approach would fulfil the goal of our project.
Things still To Do for full hydration:
Calculate the order parameters in the new AmberLipid14: Work already in progress by Samuli.Calculate the order parameters in the Ulmschneider force field: No work yet in progress.- Calculate the order parameters in the CHARMM36-UA force field: No work yet in progress.
Notes on some technical issues that came up while the results presented here were produced. These notes are probably not crucial for the goals of the nmrlipids-project, but are presented here as they might be of general interest to the project participants or to the lipid simulation community at large.
We were not able to completely reproduce the results for GAFFlipids (area per molecule, order parameters) reported in the original publication [Dickson 2012]. We tested various approaches (different lipid types, different bond constraints, different thermostats, different hydrogen masses), but the issue remains so far unsolved. It is possibly related to differences between Gromacs and Amber simulation packages.
{kind=link}
Three problems were encountered while reproducing the results of Poger et al. 2012:
- We could not reproduce the area per molecule reported in the publication.
- Contrary to what was reported in the publication, results (area per molecule and order parameters) were affected by the choice of method for calculating the electrostatic interactions: reaction field vs. PME.
- The order parameters for the headgroup and glycerol carbons of different models (Poger, Kukol, Berger) reported by Poger et al (in their Table 2) differ from the results that have been obtained in the blog.
- It turned out that Gromacs versions 4.0 and 4.5 give different results for reaction field simulations. For more details see the Redmine Bug Report.
- While changing from reaction field to PME, Poger et al. also decreased their Lennard-Jones cut-off from 1.4 nm to 1.0 nm. This could possibly compensate for the changes we observed when switching from reaction field to PME and keeping the rest of our parameters fixed.
- This was not investigated further, as none of the reported order parameter sets (neither in the blog nor in the publication) were in good agreement with the experimental values. However, it is not clear to us if the Gromacs tool g_order was maybe used by Poger et al. for calculating the headgroup order parameters—this can be problematic should the hydrogens of a single carbon experience a split, i.e., give different order parameters.
Tuesday, February 25, 2014
Accuracy of order parameter measurements
The first comment in this blog after publication of the manuscript was written by Alexander Lyubartsev, who asked a very relevant question: how accurate are the measured (and simulated) order parameters? Troughout the blog and manuscript I have assumed the absolute values of the experimentally measured order parameters to be accurate within \(\pm\) 0.02. I also stated in my guest post to the MARTINI group blog that the measured order parameters are "reliable and not sparse, and they do not depend on the technique used and the lab in which they were measured." In this post I justify my views. In addition, I will explain why the relative changes with changing conditions can be measured with higher accuracy than the absolute values.
In the ²H experiment the quadrupolar splitting \(\Delta \nu_Q\) is measured. The C-²H order parameter is
$$S_\mathrm{CD}=\frac{4}{3}\frac{h}{e²qQ} \Delta \nu_Q. (1)$$
Here \(e\) is the elementary charge, \(Q\) is the deuteron quadrupole moment and \(h\) is the Planck's constant. The parameter \(q\) is related to the largest electric field gradient and in practise its value is not known; therefore the static quadrupolar coupling constant \( \frac{e²qQ}{h}\) is defined, and its value measured for different compounds in their solid state. In C-D order parameter measurements for lipids, it is typical to use the value measured for different alkenes, \( \frac{e²qQ}{h}\)=170 kHz. The relation between order parameters and quadrupolar splittings then becomes
$$ S_\mathrm{CD}=0.00784 \times \Delta \nu_Q. $$
This relation is useful as many publications report only the quadrupolar splittings. For a review and more accurate description see the work of Seelig.
In the ¹³C NMR experiment the effective dipolar coupling \(d_\mathrm{CH}\) is measured. It must be stressed that \(d_\mathrm{CH}\) is a fundamentally different physical observable than the \(\Delta \nu_Q\) of the ²H experiment. The ¹³C-H bond order parameter is
$$ S_\mathrm{CH}=\frac{4\pi\langle r_\mathrm{CH}^3 \rangle}{\hbar \mu_0 \gamma_h \gamma_c} d_\mathrm{CH}. (2)$$
Here \(r_\mathrm{CH}\) is the C-H distance, \(\mu_0\) is the vacuum permittivity, and \(\gamma_h\) and \(\gamma_c\) are the gyromagnetic constants for ¹H and ¹³C nuclei. In contrast to Eq. (1), all the parameters in Eq. (2) are in principle known. However, for the internuclear distance only the average \(\langle r_\mathrm{CH} \rangle\) is known, not the third moment \(\langle r^3_\mathrm{CH} \rangle\). For this reason values between 20.2-22.7 kHz are used for \(\frac{\hbar \mu_0 \gamma_h \gamma_c}{4\pi\langle r_\mathrm{CH}^3 \rangle}\) [Hong et al.,Gross et al.,Dvinskikh et al.]
²H NMR and ¹³C NMR are fully independent experiments since the deuterium quadrupolar splitting \(\Delta \nu_Q\) and the effective dipolar coupling \(d_{CH}\) are different physical observables. In addition, the prefactors connecting the observables to the order parameter (Eqs. (1) and (2)) are independently measured. In ¹³C NMR further independent experiments can be performed by measuring the ¹³C dipolar couplings using different pulse sequences [Hong et al., Gross et al., Dvinskikh et al, Ferreira et al.].
The order parameter measurements are very reproducible. The results obtained for PC bilayers with different ¹³C pulse sequences and ²H measurements have been compared in Table 1 in Gross et al., Table 1 in Dvinskikh et al., and Table 1 and Fig. 3 in Ferreira et al. The order parameters measured with different techniques by independent reasearch groups are in good agreement. The results for the headgroup and glycerol region measured from samples with purified PC lipid molecules are within a maximum difference of 0.02. The larger differences (maximum 0.04) in the tables arise from the samples with E. Coli or eggPC extracts. In the acyl chains there are some carbons for which the difference is sligtly larger even for purified lipid samples, but as we are focusing on the headgroup and glycerol order parameters of purified PC lipids, we can consider these to be known with absolute accuracy of \(\pm\) 0.02.
Note. In the work by Aussenac et al. (discussed in the context of the blog here, and compared to the simulations by Högberg et al. as pointed out in here) the order parameters are measured for bicelles, not vesicles or bilayer stacks. The order parameters for bicelles are known to be smaller than the ones for vesicles by roughly a constant factor, most likely due to the bilayer orientations with respect to the magnetic field [Raffard et al., Sanders et al.]. When corrected with this constant factor, the order parameters by Aussenac et al. are also close to the ones measured for vesicles.
Clearly, the effects of different ions on the quadrupole splittings are differentiable within experimental accuracy in Fig. 1. (These changes were later shown to be consistent with the addition of different charges into the bilayer, and the electrometer concept was introduced to measure the amount of charge incorporated in the bilayer interface. [Scherer et al.]) Importantly, when the results of Fig. 1 are transformed to order parameters in Fig. 2, one can see that the numerical changes in \(S_\mathrm{CD}\) are rather small: the y-axis only spans < 0.03 units for \(\beta\) and 0.06 for \(\alpha\).
Indeed, the measured (upon addition of 1M CaCl\(_2\) concentration, the changes due NaCl are negligible) changes in the \(\beta\)-and \(\alpha\)-carbons are only \(\sim\) 0.02 and \(\sim\) 0.05, respectively.
The resolution of ¹³C experiment depends somewhat on the pulse sequence used. For example Dvinskikh et al. measured the effect of dehydration in the lipid order parameters with APM-CP pulse sequence, and changes of 0.01 were measurable. Fig. 3 shows the order parameters for \(\alpha\)-and \(\beta\)-carbons as a function of dehydration independently measured with ¹³C and ²H NMR for different PC lipids. All the results are qualitatively similar, that is, they give the same relative change with dehydration.
²H NMR and ¹³C NMR: two fundamentally different experiments
The order parameter of a C-H bond vector can be assessed with two fundamentally different NMR techniques: measuring the quadrupolar splitting from ²H spectra or measuring the dipolar splitting from ¹³C spectra.In the ²H experiment the quadrupolar splitting \(\Delta \nu_Q\) is measured. The C-²H order parameter is
$$S_\mathrm{CD}=\frac{4}{3}\frac{h}{e²qQ} \Delta \nu_Q. (1)$$
Here \(e\) is the elementary charge, \(Q\) is the deuteron quadrupole moment and \(h\) is the Planck's constant. The parameter \(q\) is related to the largest electric field gradient and in practise its value is not known; therefore the static quadrupolar coupling constant \( \frac{e²qQ}{h}\) is defined, and its value measured for different compounds in their solid state. In C-D order parameter measurements for lipids, it is typical to use the value measured for different alkenes, \( \frac{e²qQ}{h}\)=170 kHz. The relation between order parameters and quadrupolar splittings then becomes
$$ S_\mathrm{CD}=0.00784 \times \Delta \nu_Q. $$
This relation is useful as many publications report only the quadrupolar splittings. For a review and more accurate description see the work of Seelig.
In the ¹³C NMR experiment the effective dipolar coupling \(d_\mathrm{CH}\) is measured. It must be stressed that \(d_\mathrm{CH}\) is a fundamentally different physical observable than the \(\Delta \nu_Q\) of the ²H experiment. The ¹³C-H bond order parameter is
$$ S_\mathrm{CH}=\frac{4\pi\langle r_\mathrm{CH}^3 \rangle}{\hbar \mu_0 \gamma_h \gamma_c} d_\mathrm{CH}. (2)$$
Here \(r_\mathrm{CH}\) is the C-H distance, \(\mu_0\) is the vacuum permittivity, and \(\gamma_h\) and \(\gamma_c\) are the gyromagnetic constants for ¹H and ¹³C nuclei. In contrast to Eq. (1), all the parameters in Eq. (2) are in principle known. However, for the internuclear distance only the average \(\langle r_\mathrm{CH} \rangle\) is known, not the third moment \(\langle r^3_\mathrm{CH} \rangle\). For this reason values between 20.2-22.7 kHz are used for \(\frac{\hbar \mu_0 \gamma_h \gamma_c}{4\pi\langle r_\mathrm{CH}^3 \rangle}\) [Hong et al.,Gross et al.,Dvinskikh et al.]
²H NMR and ¹³C NMR are fully independent experiments since the deuterium quadrupolar splitting \(\Delta \nu_Q\) and the effective dipolar coupling \(d_{CH}\) are different physical observables. In addition, the prefactors connecting the observables to the order parameter (Eqs. (1) and (2)) are independently measured. In ¹³C NMR further independent experiments can be performed by measuring the ¹³C dipolar couplings using different pulse sequences [Hong et al., Gross et al., Dvinskikh et al, Ferreira et al.].
The order parameter measurements are very reproducible. The results obtained for PC bilayers with different ¹³C pulse sequences and ²H measurements have been compared in Table 1 in Gross et al., Table 1 in Dvinskikh et al., and Table 1 and Fig. 3 in Ferreira et al. The order parameters measured with different techniques by independent reasearch groups are in good agreement. The results for the headgroup and glycerol region measured from samples with purified PC lipid molecules are within a maximum difference of 0.02. The larger differences (maximum 0.04) in the tables arise from the samples with E. Coli or eggPC extracts. In the acyl chains there are some carbons for which the difference is sligtly larger even for purified lipid samples, but as we are focusing on the headgroup and glycerol order parameters of purified PC lipids, we can consider these to be known with absolute accuracy of \(\pm\) 0.02.
Note. In the work by Aussenac et al. (discussed in the context of the blog here, and compared to the simulations by Högberg et al. as pointed out in here) the order parameters are measured for bicelles, not vesicles or bilayer stacks. The order parameters for bicelles are known to be smaller than the ones for vesicles by roughly a constant factor, most likely due to the bilayer orientations with respect to the magnetic field [Raffard et al., Sanders et al.]. When corrected with this constant factor, the order parameters by Aussenac et al. are also close to the ones measured for vesicles.
Absolute vs. Relative accuracy
The absolute accuracy of measured order parameters is largely affected by the accuracy of the prefactor connecting the measured splittings to the order parameters (Eqs. (1) and (2)). When considering the change of an order parameter with a changing external condition (hydration, ion concentration, etc.), the prefactor can be considered to be unchanging. Therefore, the change in a given order parameter, when measured by the same people with the same technique and equipment, can be determined in a much higher accuracy than the absolute value of the order parameter. We refer to this as the relative accuracy. It is determined by the accuracy of the splitting measurement. Especially the very high spectral resolution of the ²H-measurements allows the measurement of very small order parameter changes. Let us exemplify this with a classical experiment by Akutsu et al., where the effect of different ions on the headgroup \(\alpha\)- and \(\beta\)-quadrupolar splittings was measured, see Fig. 1. |
| Figure 1. The measured quadrupolar splittings with the explanation in the original caption. Reprinted with permission from Akutsu and Seelig, Biochemistry, 20, 7366 (1981). Copyright (1981) American Chemical Society. |
 |
| Figure 2. Same data as Fig. 1. but given as order parameters instead of splittings. |
The resolution of ¹³C experiment depends somewhat on the pulse sequence used. For example Dvinskikh et al. measured the effect of dehydration in the lipid order parameters with APM-CP pulse sequence, and changes of 0.01 were measurable. Fig. 3 shows the order parameters for \(\alpha\)-and \(\beta\)-carbons as a function of dehydration independently measured with ¹³C and ²H NMR for different PC lipids. All the results are qualitatively similar, that is, they give the same relative change with dehydration.
 |
| Figure 3. Dehydration changes on \(\alpha\) and \(\beta\) order parameters measured with different methods. The data taken from Dvinskikh et al., Ulrich et al. and Bechinger et al. |
Why Simulate?
From the experiments it is not clear how large conformational and energetical changes these small changes in the order parameters indicate. This could, however, be revealed with a molecular model that reproduces the measured order parameters. We have already succesfully applied this approach to explain the measured low order parameters (and their temperature dependence) in the lamellar phase of the nonionic surfactant C\(_{12}\)E\(_5\) [Ferreira et al.]. For the lipid headgroup and glycerol region the problem is, however, that with the current force fields the order parameters from simulation models are both absolutely and relatively too far from the experimental values to be useful for interpretating the experiments.
Subscribe to:
Posts (Atom)